Clinical Ketamine Compounding Specifications requires United States Pharmacopeia (USP) or European Pharmacopoeia (EP) pharmaceutical-grade Ketamine Hydrochloride powder with a verified purity profile of ≥99.0%. Compounded solutions achieve optimal thermodynamic stability between pH 3.5 and 5.5, strictly adhering to an endotoxin limit of <0.4 EU/mg. Standard operating procedures dictate identity verification via FT-IR spectroscopy and HPLC testing, which establishes baseline stability profiles that guarantee clinical safety during downstream intravenous dilution and patient administration protocols.
Clinical Ketamine Compounding Specifications: Pharmaceutical-Grade Ketamine HCl
In the landscape of modern neuro-therapeutics and targeted pain management, precision formulation is paramount. As sublingual troches, nasal sprays, and customized intravenous admixtures increasingly define treatment-resistant depression (TRD) and chronic neuropathic pain protocols, compounding pharmacies and clinical researchers face a high-stakes scientific burden. Sourcing raw materials and manufacturing preparations demand complete clarity regarding chemical metrics, degradation pathways, and regulatory compliance indexes. This comprehensive guide evaluates the primary chemical and operational parameters governing Ketamine Hydrochloride powder (CAS 1867-66-9) formulation protocols.
1. What grade of Ketamine is used for clinical compounding?
For clinical compounding, pharmacies must strictly utilize United States Pharmacopeia (USP), European Pharmacopoeia (EP), or British Pharmacopoeia (BP) pharmaceutical-grade Active Pharmaceutical Ingredients (APIs). Raw materials designated as research-grade, industrial-grade, or chemical-grade are completely prohibited for human use due to unchecked synthetic variations and unpredictable by-product profiles.
The USP monograph dictates that the raw input material must be an ultra-pure, white crystalline powder containing not less than 98.0% and not more than 102.0% of Ketamine Hydrochloride on an anhydrous basis. To understand how these parameters shift across applications, medical buyers should consult the comprehensive USP grade ketamine hydrochloride guide. Premium global suppliers like LyfeUnit establish baseline purity margins at ≥99.8% by implementing rigorous multi-stage crystallization controls. Sourcing certified materials ensures the complete absence of harmful industrial catalysts, unreacted precursors, or unauthorized heavy metals, protecting the safety of the patient and preserving the structural integrity of the final compounded compound.
Procurement professionals looking to source specific structural variants can choose to buy ketamine powder directly through our validated B2B pipeline, or review options to buy pure ketamine crystals depending on the exact compounding machinery and dissolution protocols utilized in their laboratory setup.
2. What is the pH stability of compounded Ketamine Hydrochloride powder?
Ketamine Hydrochloride is an exceptionally robust, water-soluble molecule, yet its long-term molecular integrity in solution depends heavily on the ambient pH environment. When raw Ketamine powder is dissolved in sterile aqueous vehicles, it exhibits optimum thermodynamic stability within a pH range of 3.5 to 5.5.
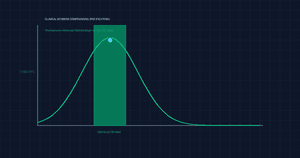
If the pH of a compounded matrix drifts significantly above 6.0, the equilibrium shifts, forcing the highly soluble hydrochloride salt into its freebase form (Ketamine base). Because the freebase variant has remarkably low water solubility, this transition leads to visual precipitation, crystalline drop-out, and localized under-dosing. Conversely, exposing the solution to extreme highly acidic conditions (pH < 2.0) risks accelerating hydrolytic cleavage, breaking the molecule down into untherapeutic secondary metabolites. Maintaining the target 3.5–5.5 bracket using appropriate buffers (such as citric acid or sodium hydroxide) prevents premature degradation, keeping the compound chemically stable across extended storage windows.
3. How do compounding pharmacies verify Ketamine powder purity?
Under standard regulatory mandates (including USP <795> for non-sterile and USP <797> for sterile compounding), formulation specialists cannot blind-trust a supplier’s label. Pharmacies must employ a dual-layer verification matrix to authenticate incoming bulk batches before release to the cleanroom floor. For a detailed breakdown of these analytical processes, review the official ketamine purity standards guide to align your internal laboratory validation checks with international benchmarks.
- Fourier-Transform Infrared Spectroscopy (FT-IR): This structural screening maps the unique molecular “fingerprint” of the incoming powder against a certified USP reference standard. It acts as an instant defense against counterfeit or cross-contaminated raw materials. Detailed molecular structures can be cross-referenced via the NCBI PubChem Ketamine Hydrochloride Database.
- High-Performance Liquid Chromatography (HPLC): HPLC serves as the quantitative benchmark for composition analysis. It measures exact enantiomeric concentrations and detects trace impurities down to fractions of a percent, verifying that the batch meets the targeted ≥99.0% specification window.
- Physical Property Metric Sweeps: Technicians cross-reference the batch by validating the precise capillary melting point (typically resting between 262°C to 263°C) alongside explicit ash residue testing.
When analyzing solid-state formulations, compounding labs often evaluate alternative structural cuts. If your facility works with non-powder variants, you can explore how to safely order clean ketamine shards to maintain uniform solubility values across larger processing batches.
4. What are the bacterial endotoxin limits for Ketamine HCl USP?
For sterile preparations, including localized epidural blocks and standard intravenous lines, keeping the compound free from pyrogens is critical. Endotoxins—lipopolysaccharides shed from gram-negative bacterial cell walls—can trigger severe systemic inflammatory responses, anaphylaxis, or septic shock if introduced into human circulation.
According to official mandates monitored by the United States Pharmacopeia (USP) Organization, Ketamine Hydrochloride injections must contain less than 0.4 USP Endotoxin Units per milligram (EU/mg) of the compound. Achieving this ultra-low baseline requires raw powder synthesis to take place inside strict, cleanroom manufacturing environments. Compounding pharmacies confirm compliance by executing Limulus Amebocyte Lysate (LAL) testing protocols on completed batches, ensuring that no pyrogenic contamination occurs during transport, storage, or final formulation steps.
5. How do you dilute Ketamine HCl powder for intravenous infusion protocols?
Reconstituting bulk powder for intravenous infusion requires flawless aseptic technique inside an ISO Class 5 laminar flow hood, ensuring the mixture remains completely sterile throughout the dilution protocol. For practitioners specializing in animal health formulations, specific adjustments should be reviewed within our USP-grade ketamine powder for veterinary compounding technical blueprint.
The process begins by converting the raw crystalline powder into a sterile concentrate. Technicians dissolve a precise mass of USP-grade powder into Sterile Water for Injection (SWFI) or 0.9% Sodium Chloride Injection (Normal Saline) to establish a standard stock concentrate (typically 10 mg/mL, 50 mg/mL, or 100 mg/mL). This solution is then passed through a sterile, pyrogen-free 0.22-micron polyethersulfone (PES) membrane filter into a sterile container to capture any potential microscopic particles.
For final clinical administration, this sterile concentrate is added to standard infusion bags filled with either 0.9% Normal Saline or 5% Dextrose in Water (D5W). A typical sub-anesthetic clinical dosage target ranges from 0.5 mg/mL to 2.0 mg/mL. The mixture must be gently inverted to ensure uniform drug distribution across the carrier matrix prior to clinical activation.
6. What is the shelf life of compounded oral Ketamine solutions?
The shelf life and Beyond-Use Date (BUD) of customized oral Ketamine formulations (such as sublingual liquids, flavored syrups, or oral drops) are determined by the storage environment and the presence of antimicrobial preservatives.
Under the conservative baseline rules of USP <795> for water-containing oral formulations, the default BUD is capped at 14 days when stored under refrigerated conditions (2°C to 8°C). However, if the formulation uses an anhydrous or low-water vehicle (such as pure storage-grade glycerin or polyethylene glycol), or incorporates robust antimicrobial preservatives (like methylparaben or sodium benzoate), this stability window can be safely extended. When supported by stability-indicating high-performance liquid chromatography validation, these configurations can maintain active drug concentrations above 90.0% for 30 to 90 days, provided they are kept in amber glass containers away from direct light and heat exposure.
Summary Comparison Matrix for Analytical Reference : Clinical Ketamine Compounding Specifications
| Compounding Variable | Target Benchmark Metric | Primary Risk Factor if Unmet |
|---|---|---|
| Chemical Grade | USP / EP / BP Compliant | Precursor toxicity & trace impurities |
| Systemic pH Window | 3.5 to 5.5 Stability Bracket | Freebase precipitation (>6.0) |
| Pyrogen Tolerance | < 0.4 EU/mg Limit | Injection fevers and systemic shock |
| Aqueous Oral BUD | 14 Days (Refrigerated) | Microbial proliferation & potency loss |
Disclaimer: The data compiled here serves as a technical, educational resource for licensed compounding pharmacies, chemical researchers, and medical distribution buyers. All compounding activities must comply strictly with national regulatory guidelines and regional pharmacy standards.



Excellent breakdown!